A selection of FELIX experiments
Note: special webpages for: [electro-optic THz detection ] [electron bunch length measurements ]
Structural identification of electron transfer dissociation products in mass spectrometry using infrared ion spectroscopy. download here Nature Communications 7 (2016) 11754.
Gas-phase conformations of small polyprolines and their fragment ions by IRMPD spectroscopy. download here International Journal of Mass Spectrometry 377 (2015) 179-187.
Laboratory infrared spectroscopy of gaseous negatively charged polyaromatic hydrocarbons. download here The Astrophysical Journal 787 (2014) 170.
Metal Ion Complexes with HisGly: Comparison with PhePhe and PheGly. download here Journal of Physical Chemistry A 117 (2013) 5335–5343.
Isomer-Selective Detection of Hydrogen-Bond Vibrations in the Protonated Water Hexamer. download here Journal of the American Chemical Society 135 (2013) 8266-8273
Infrared multiple photon dissociation (IRMPD) spectroscopy of oxazine dyes. download here Physical Chemistry Chemical Physics 15 (2013) 5049-5056.
Metal ion binding to peptides: Oxygen or nitrogen sites? download here International Journal of Mass Spectrometry 330-332 (2012) 71-77.
Peptide Bond Tautomerization Induced by Divalent Metal Ions: Characterization of the Iminol Configuration. download here Angewandte Chemie International Edition 51 (2012) 4591–4593.
Non-Equilibrium Isomer Distribution of the Gas-Phase Photoactive Yellow Protein Chromophore. download here click here Journal of Physical Chemistry Letters 3 (2012) 2259-2263.
Stability of Gas-Phase Tartaric Acid Anions Investigated by Quantum Chemistry, Mass Spectrometry, and Infrared Spectroscopy. download here Journal of Physical Chemistry A 116 (2012) 4789–4800.
Examination of the Coordination Sphere of AlIII in Trifluoromethyl-Heteroarylalkenolato Complex Ions by Gas-Phase IRMPD Spectroscopy and Computational Modelling. download here ChemPhysChem 13 (2012) 2037-2045.
Tweezer-like Complexes of Crown Ethers with Divalent Metals: Probing Cation-Size-Dependent Conformations by Vibrational Spectroscopy in the Gas Phase. download here ChemPlusChem 77 (2012) 118-123.
Mid-infrared spectroscopy of molecular ions in helium nanodroplets. download here Journal of Chemical Physics 136 (2012) 044305.
Structural Elucidation of Biological and Toxicological Complexes: Investigation of Monomeric and Dimeric Complexes of Histidine with Multiply Charged Transition Metal (Zn and Cd) Cations using IR Action Spectroscopy. download here Journal of Physical Chemistry B 115 (2011) 12648–12661.
Structure and Reactivity of the N-Acetyl-Cysteine Radical Cation and Anion: Does Radical Migration Occur? Victor Ryzhov download here Journal of The American Society for Mass Spectrometry 22 (2011) 1794-1803.
Direct evidence for the ring opening of monosaccharide anions in the gas phase: photodissociation of aldohexoses and aldohexoses derived from disaccharides using variable-wavelength infrared irradiation in the carbonyl stretch region. download here Carbohydrate Research 346 (2011) 2469-2481.
Infrared spectra of the protonated neurotransmitter histamine: competition between imidazolium and ammonium isomers in the gas phase. download here Physical Chemistry Chemical Physics 13 (2011) 15644-15656.
Infrared multiple photon dissociation action spectroscopy of sodiated uracil and thiouracils: Effects of thioketo-substitution on gas-phase conformation. International Journal of Mass Spectrometry
Vibrational study of isolated 18-crown-6 ether complexes with alkaline-earth metal cations. International Journal of Mass Spectrometry
IR Spectroscopy of Isolated Neutral and Protonated Adenine and 9-Methyladenine. download here Chem. Phys. Chem. 12 (2011) 1921-1927.
Isomer Population Analysis of Gaseous Ions From Infrared Multiple Photon Dissociation Kinetics. Evan R. Williams download here Journal of Physical Chemistry A 115 (2011) 2745-2751.
Gas Phase Infrared Multiple Photon Dissociation Spectra of Positively Charged Sodium Bis(2-ethylhexyl)sulfosuccinate Reverse Micelle-like Aggregates. download here Journal of Physical Chemistry B 115 (2011) 2282-2286.
Time-Resolved Holography with Photoelectrons. M.J.J. Vrakking download here Science 331 (2011) 61-64.
Infrared Spectra of Protonated Neurotransmitters: dopamine download here Physical Chemistry Chemical Physics 13 (2011) 2815-2823.
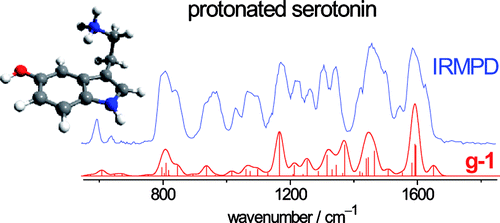
Infrared Spectra of Protonated Neurotransmitters: Serotonin download here Journal of Physical Chemistry A 114 (2010) 13268-13276.
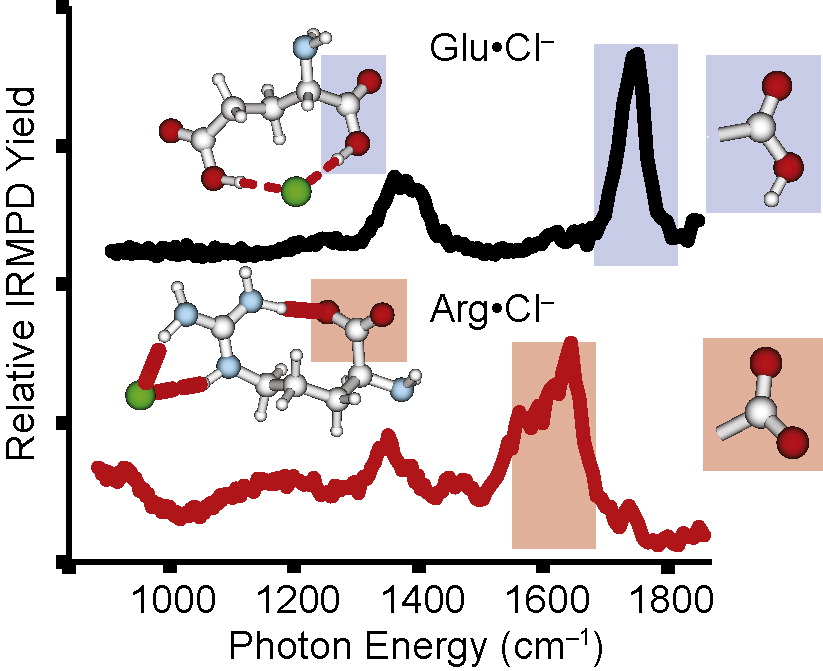
Effects of Anions on the Zwitterion Stability of Glu, His and Arg Investigated by IRMPD Spectroscopy and Theory Evan R. Williams download here International Journal of Mass Spectrometry 297 (2010) 116-123.
Coordination of Trivalent Metal Cations to Peptides: Results from IRMPD Spectroscopy and Theory Evan R. Williams download here Journal of Physical Chemistry A 114 (2010) 854-860.
Above-threshold ionization in a strong dc electric field
Physical Review A 78 (2008) 013413. URL Link: http://link.aps.org/abstract/PRA/v78/e013413
Spin Relaxation by Transient Monopolar and Bipolar Optical Orientation download here Phys. Rev. Lett. 96, 096603 (2006)
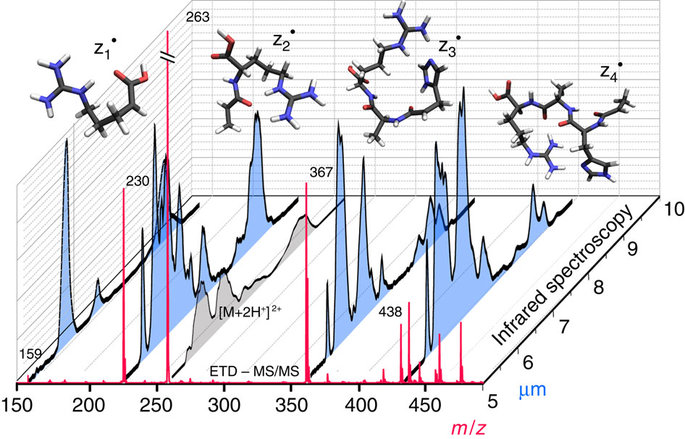 Structural identification of electron transfer dissociation products in mass spectrometry using infrared ion spectroscopy.
Structural identification of electron transfer dissociation products in mass spectrometry using infrared ion spectroscopy.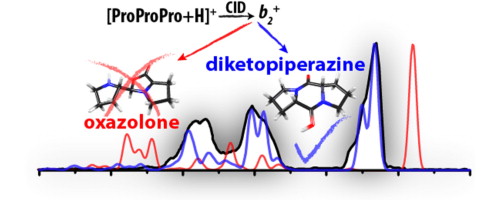 Gas-phase conformations of small polyprolines and their fragment ions by IRMPD spectroscopy.
Gas-phase conformations of small polyprolines and their fragment ions by IRMPD spectroscopy. Laboratory infrared spectroscopy of gaseous negatively charged polyaromatic hydrocarbons.
Laboratory infrared spectroscopy of gaseous negatively charged polyaromatic hydrocarbons. Metal Ion Complexes with HisGly: Comparison with PhePhe and PheGly.
Metal Ion Complexes with HisGly: Comparison with PhePhe and PheGly. Isomer-Selective Detection of Hydrogen-Bond Vibrations in the Protonated Water Hexamer.
Isomer-Selective Detection of Hydrogen-Bond Vibrations in the Protonated Water Hexamer.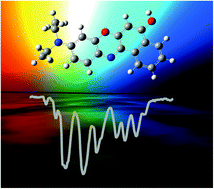 Infrared multiple photon dissociation (IRMPD) spectroscopy of oxazine dyes.
Infrared multiple photon dissociation (IRMPD) spectroscopy of oxazine dyes.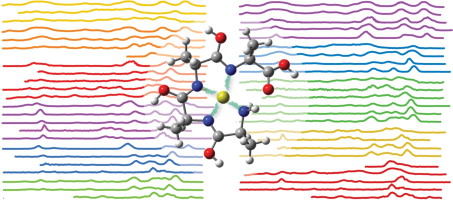 Metal ion binding to peptides: Oxygen or nitrogen sites?
Metal ion binding to peptides: Oxygen or nitrogen sites?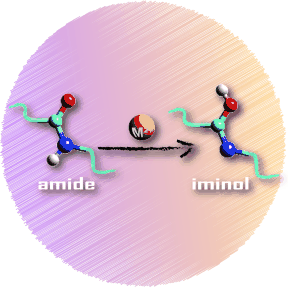 Peptide Bond Tautomerization Induced by Divalent Metal Ions: Characterization of the Iminol Configuration.
Peptide Bond Tautomerization Induced by Divalent Metal Ions: Characterization of the Iminol Configuration.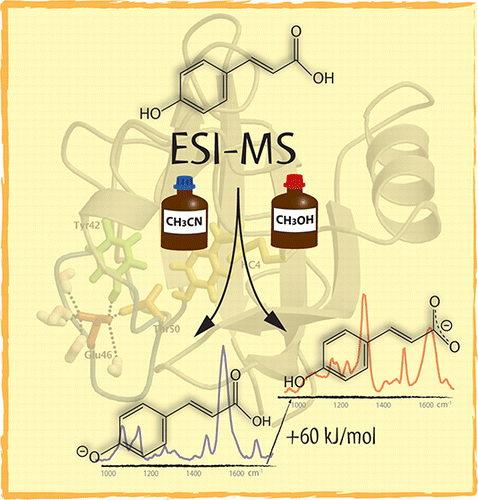 Non-Equilibrium Isomer Distribution of the Gas-Phase Photoactive Yellow Protein Chromophore.
Non-Equilibrium Isomer Distribution of the Gas-Phase Photoactive Yellow Protein Chromophore.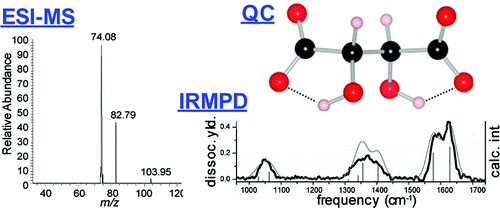 In an effort to understand the chemical factors that stabilize dianions, experimental and theoretical studies on the stability of the tartrate dianion were performed. Quantum chemical calculations at the coupled cluster level reveal only a metastable state with a possible decomposition pathway (O2C–CH(OH)–CH(OH)–CO2)2– --> (O2C–CH(OH)–CH(OH))•– + CO2 + e– explaining the observed gas-phase instability of this dianion. Further theoretical data were collected for the bare dianion, this molecule complexed to water, sodium, and a proton, in both the meso and l forms as well as for the uncomplexed radical anion and neutral diradical. The calculations suggest that the l-tartrate dianion is more thermodynamically stable than the dianion of the meso stereoisomer and that either dianion can be further stabilized by association with a separate species that can help to balance the charge of the molecular complex. Mass spectrometry was then used to measure the energy needed to initiate collisionally induced dissociation of the racemic tartrate dianion and for the proton and sodium adducts of both the racemic and meso form of this molecule. Infrared action spectra of the dianion stereoisomers complexed with sodium were also acquired to determine the influence of the metal ion on the vibrations of the dianions and validate the computationally predicted structures. These experimental data support the theoretical conclusions and highlight the instability of the bare tartrate dianion. From the experimental work, it could also be concluded that the pathway leading to dissociation is under kinetic control because the sodium adduct of the racemic stereoisomer dissociated at lower collisional energy, although it was calculated to be more stable, and that decomposition proceeded via C–C bond dissociation as computationally predicted. Taken together, these data provide insight into the gas-phase stability of the tartrate dianion and highlight the role of adducts in stabilizing this species. © 2012 American Chemical Society.
In an effort to understand the chemical factors that stabilize dianions, experimental and theoretical studies on the stability of the tartrate dianion were performed. Quantum chemical calculations at the coupled cluster level reveal only a metastable state with a possible decomposition pathway (O2C–CH(OH)–CH(OH)–CO2)2– --> (O2C–CH(OH)–CH(OH))•– + CO2 + e– explaining the observed gas-phase instability of this dianion. Further theoretical data were collected for the bare dianion, this molecule complexed to water, sodium, and a proton, in both the meso and l forms as well as for the uncomplexed radical anion and neutral diradical. The calculations suggest that the l-tartrate dianion is more thermodynamically stable than the dianion of the meso stereoisomer and that either dianion can be further stabilized by association with a separate species that can help to balance the charge of the molecular complex. Mass spectrometry was then used to measure the energy needed to initiate collisionally induced dissociation of the racemic tartrate dianion and for the proton and sodium adducts of both the racemic and meso form of this molecule. Infrared action spectra of the dianion stereoisomers complexed with sodium were also acquired to determine the influence of the metal ion on the vibrations of the dianions and validate the computationally predicted structures. These experimental data support the theoretical conclusions and highlight the instability of the bare tartrate dianion. From the experimental work, it could also be concluded that the pathway leading to dissociation is under kinetic control because the sodium adduct of the racemic stereoisomer dissociated at lower collisional energy, although it was calculated to be more stable, and that decomposition proceeded via C–C bond dissociation as computationally predicted. Taken together, these data provide insight into the gas-phase stability of the tartrate dianion and highlight the role of adducts in stabilizing this species. © 2012 American Chemical Society.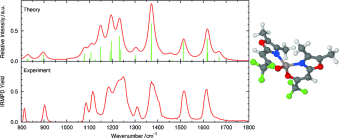 Lisa Brückmann, Wieland Tyrra, Sanjay Mathur, Giel Berden, Jos Oomens, Anthony J. H. M. Meijer and Mathias Schäfer
Lisa Brückmann, Wieland Tyrra, Sanjay Mathur, Giel Berden, Jos Oomens, Anthony J. H. M. Meijer and Mathias Schäfer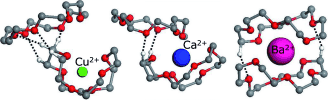 Tweezer-like Complexes of Crown Ethers with Divalent Metals: Probing Cation-Size-Dependent Conformations by Vibrational Spectroscopy in the Gas Phase.
Tweezer-like Complexes of Crown Ethers with Divalent Metals: Probing Cation-Size-Dependent Conformations by Vibrational Spectroscopy in the Gas Phase. Mid-infrared spectroscopy of molecular ions in helium nanodroplets.
Mid-infrared spectroscopy of molecular ions in helium nanodroplets.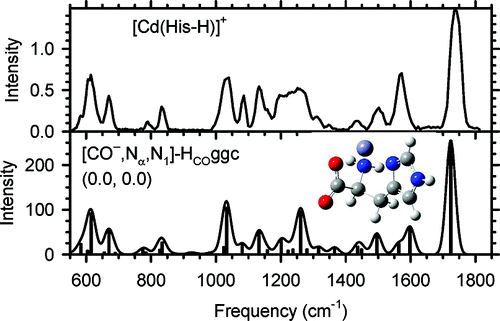 Structural Elucidation of Biological and Toxicological Complexes: Investigation of Monomeric and Dimeric Complexes of Histidine with Multiply Charged Transition Metal (Zn and Cd) Cations using IR Action Spectroscopy.
Structural Elucidation of Biological and Toxicological Complexes: Investigation of Monomeric and Dimeric Complexes of Histidine with Multiply Charged Transition Metal (Zn and Cd) Cations using IR Action Spectroscopy.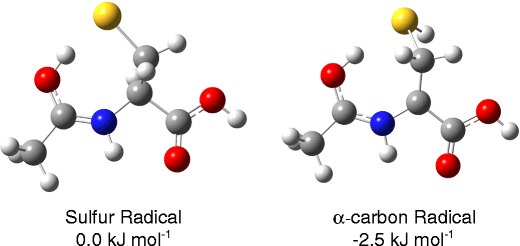 Structure and Reactivity of the N-Acetyl-Cysteine Radical Cation and Anion: Does Radical Migration Occur?
Structure and Reactivity of the N-Acetyl-Cysteine Radical Cation and Anion: Does Radical Migration Occur?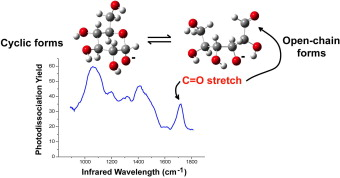 Direct evidence for the ring opening of monosaccharide anions in the gas phase: photodissociation of aldohexoses and aldohexoses derived from disaccharides using variable-wavelength infrared irradiation in the carbonyl stretch region.
Direct evidence for the ring opening of monosaccharide anions in the gas phase: photodissociation of aldohexoses and aldohexoses derived from disaccharides using variable-wavelength infrared irradiation in the carbonyl stretch region.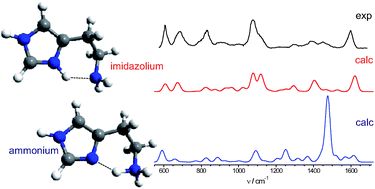 Infrared spectra of the protonated neurotransmitter histamine: competition between imidazolium and ammonium isomers in the gas phase.
Infrared spectra of the protonated neurotransmitter histamine: competition between imidazolium and ammonium isomers in the gas phase.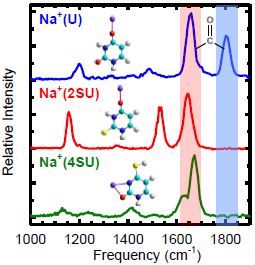 Infrared multiple photon dissociation action spectroscopy of sodiated uracil and thiouracils: Effects of thioketo-substitution on gas-phase conformation.
Infrared multiple photon dissociation action spectroscopy of sodiated uracil and thiouracils: Effects of thioketo-substitution on gas-phase conformation.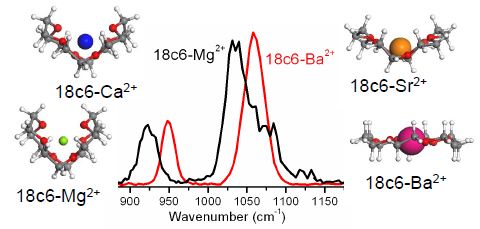 Vibrational study of isolated 18-crown-6 ether complexes with alkaline-earth metal cations.
Vibrational study of isolated 18-crown-6 ether complexes with alkaline-earth metal cations. IR Spectroscopy of Isolated Neutral and Protonated Adenine and 9-Methyladenine.
IR Spectroscopy of Isolated Neutral and Protonated Adenine and 9-Methyladenine. Isomer Population Analysis of Gaseous Ions From Infrared Multiple Photon Dissociation Kinetics.
Isomer Population Analysis of Gaseous Ions From Infrared Multiple Photon Dissociation Kinetics. Gas Phase Infrared Multiple Photon Dissociation Spectra of Positively Charged Sodium Bis(2-ethylhexyl)sulfosuccinate Reverse Micelle-like Aggregates.
Gas Phase Infrared Multiple Photon Dissociation Spectra of Positively Charged Sodium Bis(2-ethylhexyl)sulfosuccinate Reverse Micelle-like Aggregates. Time-Resolved Holography with Photoelectrons.
Time-Resolved Holography with Photoelectrons.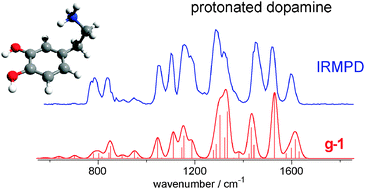
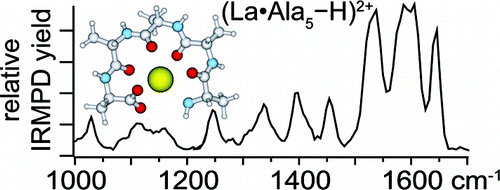 Abstract: Structures of trivalent lanthanide metal cations La3+, Ho3+, and Eu3+ with deprotonated Alan (n = 2-5) or Leu-enk (Tyr-Gly-Gly-Phe-Leu) are investigated with infrared multiple photon dissociation (IRMPD) spectroscopy between 900 and 1850 cm-1 and theory. In all of these complexes, a salt bridge is formed in which the metal cation coordinates to the carboxylate group of the peptide, resulting in a limited conformational space and many sharp IRMPD spectral bands. The IRMPD spectra clearly indicate that all carbonyl groups solvate the metal cation in each of the Alan complexes. Due to strong vibrational coupling between the carbonyl groups, a sharp, high-energy amide I band due to in-phase stretching of all of the amide carbonyl groups bound to the metal cation is observed that is separated by 50 cm-1 from a strong, lower-energy amide I band. This extent of carbonyl coupling, which is sometimes observed in condensed-phase peptide and protein IR spectroscopy, has not been reported in IRMPD spectroscopy studies of other cationized peptide complexes. Intense bands due to carbonyl groups not associated with the metal cation are observed for Leu-enk complexes, indicating that a side chain group, such as the Tyr or Phe aromatic ring, prevents complete carbonyl coordination of the metal cation. Substitution of smaller lanthanide cations for La3+ in these peptide complexes results only in minor structural changes consistent with the change in metal cation size. These are the first IRMPD spectra reported for lanthanide metal cationized peptides, and comparison to previously reported protonated and alkali metal or alkaline earth metal cationized peptide complexes reveals many trends consistent with the higher charge state of the lanthanide cations.
© 2010 American Chemical Society.
Abstract: Structures of trivalent lanthanide metal cations La3+, Ho3+, and Eu3+ with deprotonated Alan (n = 2-5) or Leu-enk (Tyr-Gly-Gly-Phe-Leu) are investigated with infrared multiple photon dissociation (IRMPD) spectroscopy between 900 and 1850 cm-1 and theory. In all of these complexes, a salt bridge is formed in which the metal cation coordinates to the carboxylate group of the peptide, resulting in a limited conformational space and many sharp IRMPD spectral bands. The IRMPD spectra clearly indicate that all carbonyl groups solvate the metal cation in each of the Alan complexes. Due to strong vibrational coupling between the carbonyl groups, a sharp, high-energy amide I band due to in-phase stretching of all of the amide carbonyl groups bound to the metal cation is observed that is separated by 50 cm-1 from a strong, lower-energy amide I band. This extent of carbonyl coupling, which is sometimes observed in condensed-phase peptide and protein IR spectroscopy, has not been reported in IRMPD spectroscopy studies of other cationized peptide complexes. Intense bands due to carbonyl groups not associated with the metal cation are observed for Leu-enk complexes, indicating that a side chain group, such as the Tyr or Phe aromatic ring, prevents complete carbonyl coordination of the metal cation. Substitution of smaller lanthanide cations for La3+ in these peptide complexes results only in minor structural changes consistent with the change in metal cation size. These are the first IRMPD spectra reported for lanthanide metal cationized peptides, and comparison to previously reported protonated and alkali metal or alkaline earth metal cationized peptide complexes reveals many trends consistent with the higher charge state of the lanthanide cations.
© 2010 American Chemical Society.
 B.N. Murdin, K. Litvinenko, D.G. Clarke, C.R. Pidgeon, P. Murzyn, P.J. Phillips, D. Carder, G. Berden, B. Redlich, A.F.G. van der Meer, S. Clowes, J.J. Harris, L.F. Cohen, T. Ashley and L. Buckle
B.N. Murdin, K. Litvinenko, D.G. Clarke, C.R. Pidgeon, P. Murzyn, P.J. Phillips, D. Carder, G. Berden, B. Redlich, A.F.G. van der Meer, S. Clowes, J.J. Harris, L.F. Cohen, T. Ashley and L. Buckle