High resolution UV spectroscopy of aromatic
molecules
In an ongoing research project we aim at a measurement of
the electronic spectra of large organic molecules with the highest possible
spectral resolution. The combination of an intra-cavity frequency
doubled cw ring dye laser with a carefully designed molecular beam
machine and a sensitive laser induced fluorescence detection set-up
has yielded a molecular beam spectrometer with a spectral resolution of
1 part in 10^8 over the whole near-UV range of the spectrum.
Rotationally resolved electronic spectra of increasingly challenging
polyatomic molecules as well as of their van der Waals and hydrogen
bonded complexes are measured in this apparatus, and their
geometrical structures are deduced. In the near future
double-resonance techniques, using microwave or infrared preselection
of one ro-vibrational level which is then probed by the tunable UV laser,
will be employed to expand the applicability of this spectrometer to even
larger molecules.
The combination of a supersonic molecular beam expansion and a narrow
band UV laser is a powerful tool in experimental molecular spectroscopy.
It can provide detailed information about the dynamics and structure of
molecules and molecular complexes in both their ground and
electronically excited states. Expanding volatilized organic molecules
seeded in a carrier gas produces a cooling of the vibrational and
rotational degrees of freedom. The advantage for high resolution
spectroscopy is two-fold. On the one hand, only the lowest rotational
and vibrational levels in the electronic ground state are populated,
leading to less congested excitation spectra. On the other hand, the
low internal temperatures permit the stabilization of structural
variants (tautomers or conformers) and the stabilization of molecular
clusters (van der Waals and hydrogen bonded complexes).
Analysis of rotationally resolved laser induced fluorescence (LIF)
spectra provides the molecular constants in both the ground and the
electronically excited state. These constants are directly related to
the geometrical structures in both states, giving access to
information about intramolecular bond lengths, and in the case of a
molecular complexes, intermolecular bond lengths and their changes upon
excitation.
Unfortunately, the number of molecular constants is too small for the
determination of the complete molecular structure. Consider, for
example, a molecule with N atoms that can be described with an
asymmetric rigid rotor Hamiltonian. There are only three rotational
constants A, B and C available, while there are 3N-3 unknown parameters.
Recording the rotationally resolved LIF spectrum of an isotopically
substituted molecule can give extra information to determine the
position of the substituted atom via Kraitchman's equations. Obviously,
in the case of a molecule containing for example 30 atoms, determination
of all atomic positions would be a very tedious process. This method is
used if only a particular part of the molecule is interesting, such as
the NH2 group in 1-aminonaphthalene.
If the molecule consists of parts with a well-known structure, the
rotational constants of the entire molecule contain enough information
to determine its structure. An example is
triphenylamine (TPA), a
molecule which consists of a nitrogen atom with three phenyl groups
attached to it. Since the structure of each phenyl group is known,
there are only a few unknown parameters left, which are related to the
relative orientation of the phenyl groups. Therefore, it is possible to
determine the structure of the entire molecule. In all other cases the
molecular constants have to be compared with other (related) molecules
or with ab initio calculations to provide information about the
structure. This last comparison is very important; it is a sensitive
check of the methods of calculation that are frequently used for
providing a large amount of information about molecular properties.
In addition to the molecular constants, one can determine the
orientation of the electronic transition moment vector in the molecular
frame from the high resolution spectrum. This vector provides
information about the direction of the electronic charge migration or
displacement that occurs during the transition. It is therefore related
to the probability distribution functions in the involved electronic
states. Furthermore, from a deconvolution of the rotational line shape,
the natural linewidth of the molecular transition can be obtained which
gives the lifetime of the excited state.
High resolution UV spectroscopy also can provide information about
interactions between electonic states. As an example, fluorescence
excitation spectra can be perturbed by a coupling with `dark' states
(ISC, intersystem crossing). The excitation spectrum of pyrazine
contains many more lines than expected, owing to a coupling between
single rovibronic levels of the S1 state with many quasi-isoenergetic
rovibronic levels of the lowest triplet state (T1). Similar
perturbations have been observed in the spectra of pyrimidine,
sym-triazine and acetylene.
Another interesting interaction is caused by the coupling of an
internal hindered rotation with the overall rotation of the molecule.
Full analysis of the spectra can provide values for the
barrier heights in the ground state and the excited
state. The extent of complexity of the spectra
depends strongly on the barrier heights, the direction of the internal
rotation axis with respect to the overall inertial axis, and the
(optical) selection rules (type of transition). In
phenol, every
rotational line is split into two barely resolved components due to
the torsion of the hydroxyl group around the C--O bond.
More complex is methylindole: internal rotation of the methyl
group leads to a spectrum which consists of two bands (A and E lines),
in which the E lines are further split by a Ka-dependent
interaction.
A number of high resolution UV experiments have been performed covering
most of the aforementioned topics. The spectrometer consists of a
molecular beam apparatus and an intracavity frequency-doubled
continuous-wave (cw) ring dye laser (265--340 nm), and has a spectral
resolution of 1 part in 10^8. The studied molecular systems differ
largely in size: from a single molecule containing 12 atoms
(phenol) to
a cluster containing 41 atoms (the van der Waals complex of
1-cyanonaphthalene and triethylamine).
Rotationally resolved fluorescence excitation spectra are obtained
using a narrow bandwidth UV laser system and a molecular beam apparatus.
The sample is heated in a quartz oven to bring it into the gas phase,
seeded in 0.2-1.0 bar argon, and expanded through a nozzle with a
diameter of 0.15 mm. The nozzle is kept at a slightly higher
temperature to prevent condensation of the sample in the orifice. The
molecular beam is skimmed twice in a differential pumping system and
is crossed perpendicularly with a UV laser beam at about 30 cm from the
nozzle.
UV radiation with a bandwidth of 3 MHz is generated by intracavity
frequency doubling in a single frequency ring dye laser. Typically
0.1--5 mW of tunable radiation can be obtained. For relative frequency
calibration a temperature stabilized Fabry-Perot interferometer is used
with a free spectral range of 75 MHz. For absolute frequency
calibration, the iodine absorption spectrum is recorded simultaneously.
The total undispersed fluorescence is imaged on a photomultiplier
connected to a photon counting system interfaced with a computer.

Both vibrationally and rotationally resolved spectra of the S1 <-- S0 transition
in jet-cooled triphenylamine (TPA) around 340-320 nm are reported. Medium resolution
spectra (0.5-1.0 cm-1 resolution) are recorded using (1+1)-Resonance Enhanced
Multi Photon Ionization (REMPI) with mass selective Time-Of-Flight (TOF) detection
in a pulsed molecular beam apparatus. The origin of the S1 <-- S0 transition
is at 29520.7 cm-1, higher than halfway to the ionization potential (IP) found
at 6.89 eV. A vibrational progression in the symmetric torsion mode (114 cm-1)
as well as in the symmetric C--N stretching mode (280 cm-1) is observed in the
electronic spectra. The spectrum of the most abundant isomer of the TPA--Ar (TPA--Kr)
complexes is blue-shifted by 211 cm-1 (216 cm-1) with respect to the spectrum
of the free TPA molecule. High resolution spectra are recorded using Laser Induced
Fluorescence (LIF) in a cw molecular beam apparatus. Individual rotational transitions
are resolved and the spectrum shows unambiguously that TPA is a symmetric top
molecule. The spectrum of the blue-shifted TPA--Ar isomer is the spectrum of a
symmetric top molecule as well, and therefore the Ar atom has to be located on
the C3 symmetry axis, either on top of or underneath the umbrella formed by the
phenyl rings. It appears that when Ar or Kr forms a complex with TPA, the first
Ar, Kr, atom goes preferentially in a position on the C3 symmetry axis of TPA,
a position which causes an abnormal blue-shift of the spectrum. With the first
rare gas atom located in this special position, the second rare gas atom is forced
into a `normal' position, i.e. above one of the phenyl-rings, causing a normal
red-shift with respect to the TPA--Ar complex. (ref)
The rotationally resolved fluorescence excitation spectrum of the 0-0 band in
the S1 <-- S0 transition of 1-cyanonaphthalene (CNN), at 318 nm, has been recorded
using laser induced fluorescence in a molecular beam apparatus. This band exhibits
pure a-type character and consists of about 600 lines at a rotational temperature
of 2.5 K, each with a linewidth of 17 MHz. A microwave-ultraviolet double resonance
experiment on the 0-0 band of CNN has been performed to verify the rotational
assignments of the fluorescence excitation spectrum and to obtain more accurate
rotational constants in both the ground and electronically excited states. The
band origin is at 31411.114 ±0.003 cm-1 and the rotational constants are
(in MHz) A''=1478.65(2), B''=956.75(1), C''=580.989(7), A'-A''=-21.363(9), B'-B''=-13.305(5),
and C'-C''=-8.167(2).(ref)
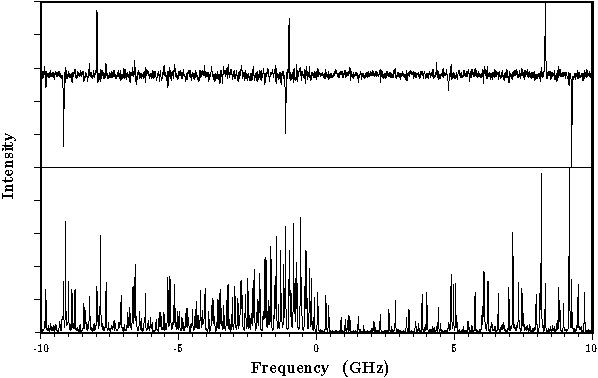
The rotationally resolved fluorescence excitation spectrum of the 0-0 band in
the S1 <-- S0 transition, at 318 nm, of the 1-cyanonaphthalene/triethylamine
van der Waals complex has been recorded using laser induced fluorescence in a
molecular beam apparatus. This spectrum could be fitted to a pure a-type band.
From the rotational constants a T-shaped geometry could be deduced.(ref)
The high resolution fluorescence excitation spectrum of the origin band of the
S1 <-- S0 transition of 1-aminonaphthalene (1AN) has been recorded. It was
found that this band is predominantly b-axis polarized, in contrast with other
(previously measured) 1-substituted naphthalenes, which are a-axis polarized.
Thirteen vibronic bands of 1AN were also examined at high resolution. The rotational
constants, the inertial defects, and the band polarizations vary significantly
from band to band. Similar experiments have been performed on eight deuterated
isotopomers. A comparison of the results obtained for these isotopomers with those
of the corresponding bands in protonated 1AN makes possible the determination
of the center-of-mass coordinates of the amino hydrogen atoms. In the zero point
vibrational level (ZPL) of the S0 state, the out-of-plane positions of the amino
hydrogens are inequivalent; the `inside' hydrogen is located 0.49(8) Angstrom
out-of-plane, the `outside' hydrogen only 0.24(17) Angstrom. In the ZPL of the
S1 state, 1AN is quasi-planar.(ref)
The rotationally resolved excitation spectrum of the 0-0 band of the S1 <--
S0 transition in 2H-benzotriazole, at 286.4 nm, is obtained by using laser induced
fluorescence spectroscopy in a molecular beam. From this pure b-type spectrum,
the rotational constants in the ground state and the electronically excited state
are determined. The rotational lines are strongly broadened due to the short lifetime
which is determined to be around 1.1 ns.(ref)
Rotationally resolved laser induced fluorescence excitation spectra of the S1
(1Lb) <-- S0 origin bands of indole, indazole, and benzimidazole have been
measured. From these spectra, the rotational constants in both electronic states
have been determined. The 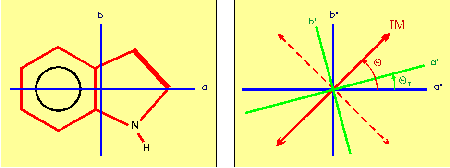 spectra of all three molecules exhibit `anomalous' rotational
line intensities. These intensity perturbations are a result of the reorientation,
upon electronic excitation, of the inertial axes of the molecule. Intensity analysis
of the rotational lines yielded information about the inertial axis reorientation,
and the direction of the transition moment vector for each molecule.(ref)
spectra of all three molecules exhibit `anomalous' rotational
line intensities. These intensity perturbations are a result of the reorientation,
upon electronic excitation, of the inertial axes of the molecule. Intensity analysis
of the rotational lines yielded information about the inertial axis reorientation,
and the direction of the transition moment vector for each molecule.(ref)
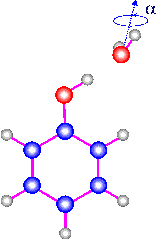 The S1 <-- S0 0-0 transitions of phenol and the hydrogen bonded phenol-water
cluster have been studied by high resolution fluorescence excitation spectroscopy.
All lines in the monomer spectrum are split by 56±4 MHz due to the internal
rotation of the --OH group about the a-axis. The barrier for this internal motion
is determined in the ground and excited states; V_2''=1215 cm-1, and V_2'=4710
cm-1. The rotational constants for the monomer in the ground state are in agreement
with those reported in microwave studies. The excited state rotational constants
were found to be A'=5313.7 MHz, B'=2620.5 MHz, and C'=1756.08 MHz. The region
of the redshifted 0-0 transition of phenol-water shows two distinct bands which
are 0.85 cm-1 apart. Their splitting arises from a torsional motion which interchanges
the two equivalent H-atoms in the H2O moiety of the cluster. This assignment was
confirmed by spin statistical considerations. Both bands could be fit to rigid
rotor Hamiltonians. Due to the interaction between the overall rotation of the
entire cluster and the internal rotation, both bands have different rotational
constants. They show that V_2' < V_2'', and that the internal rotation axis
is nearly parallel to the a-axis of the cluster. If it is assumed that the structure
of the rotor part does not change upon electronic excitation, the internal motion
becomes simply a rotation of the water molecule around its symmetry axis. Assuming
this motion, barriers of 180 cm-1 and 130 cm-1 could be estimated for the S0 and
S1 states, respectively. The analysis of the rotational constants of the cluster
yielded an O--O distance of the hydrogen bond of 2.93 angstrom in the ground state
and 2.89 angstrom in the electronically excited state. In the equilibrium structure
of the cluster, the plane containing phenol bisects the plane of the water molecule.(ref)
The rotationally resolved UV excitation spectra of the S1(1Lb)<--S0 origin bands
of 3-methylindole and 5-methylindole have been measured and analyzed. As a result
of an internal rotation of the methyl group, each spectrum consists of rotational
lines of overlapping 0a1<--0a1 and 0e<--0e torsional transitions. Like indole,
3-methylindole and 5-methylindole undergo axis reorientation upon electronic excitation.
The Hamiltonian used to describe all observed spectral includes a pure rotational
part, a pure torsional part, and terms describing the interaction between the
internal rotation and the overall rotation. It also accounts for the axis reorientation
effect. Values for the barrier heights of the methyl torsion, the angle of the
methyl top axis with the inertial axes, and the rotational constants are obtained
for both the S0 and the S1 state. From an analysis of the intensities of the rotational
transitions, the direction of the transition moment and the axis reorientation
angle are obtained. Due to quantum interference effects in the 5-methylindole
spectrum the sign of these angles could be determined. (ref)
The electronic transitions of o-fluorophenol situated at 36799.382 cm-1 and 36906.710
cm-1, denoted the A and B bands, respectively, have been investigated by high
resolution fluorescence excitation spectroscopy. Hole burning studies together
with the high resolution spectroscopy results show that both bands originate in
the same ground state and can be fitted to the rotational constants of the cis
isomer. The rotational constants for the excited states are found to be A = 3231.795
MHz, B = 2207.92 MHz and C = 1313.97 MHz for the A band and A = 3226.945 MHz,
B = 2211.24 MHz and C = 1321.03 MHz for the B band. The planarity of the ground
state is lost upon electronic excitation, which enhances the activity of an out-of-plane
vibration. The A and B band transitions arise from excitations to respectively
the zero and first overtone levels in the double-minimum potential of this out-of-plane
vibration, which shows similarities to the so-called butterfly mode observed in
other benzene derivatives.(ref)
The rotationally resolved fluorescence excitation spectrum of the 0-0 band in
the S1 <-- S0 transition of 4-aminobenzonitrile (ABN) was recorded, at 299
nm, by using laser induced fluorescence in a molecular beam apparatus. This spectrum
exhibits pure b-type character, which indicates that the electronic transition
moment vector is oriented along the short molecular axis. The rotational constants
of the S0 and S1 states were determined. In addition, the rotationally resolved
fluorescence excitation spectra of two vibronic bands in the S1 state, at 807
and 816 cm-1, were recorded. The molecular structure of the ABN molecule is discussed
by comparing the rotational constants and the inertial defects.(ref)
High resolution ultraviolet spectroscopy has been used to investigate the rotationally
resolved excitation spectrum of the first singlet-singlet transition in the benzoic
acid dimer. The measured spectrum consists of two overlapping components. The
corresponding lines in the two components are shown to originate in different
levels of the ground state potential separated by a tunneling splitting produced
by concerted proton exchange between the two subunits forming the dimer. The frequency
separation between the two components is equal to the difference between the tunneling
splittings in the ground and the excited electronic state. This frequency separation
is found to be 1107±7 MHz. From the analysis, it is estimated that the barrier
for proton tunneling changes by about 20% upon electronic excitation. The structure
of the dimer in the ground state is determined to be linear, while in the excited
S1 state it is slightly bent (3.4°±1.7°).(ref)
The S1 <-- S0 0-0 transitions of phenol and the hydrogen bonded phenol-water
cluster have been studied by high resolution fluorescence excitation spectroscopy.
All lines in the monomer spectrum are split by 56±4 MHz due to the internal
rotation of the --OH group about the a-axis. The barrier for this internal motion
is determined in the ground and excited states; V_2''=1215 cm-1, and V_2'=4710
cm-1. The rotational constants for the monomer in the ground state are in agreement
with those reported in microwave studies. The excited state rotational constants
were found to be A'=5313.7 MHz, B'=2620.5 MHz, and C'=1756.08 MHz. The region
of the redshifted 0-0 transition of phenol-water shows two distinct bands which
are 0.85 cm-1 apart. Their splitting arises from a torsional motion which interchanges
the two equivalent H-atoms in the H2O moiety of the cluster. This assignment was
confirmed by spin statistical considerations. Both bands could be fit to rigid
rotor Hamiltonians. Due to the interaction between the overall rotation of the
entire cluster and the internal rotation, both bands have different rotational
constants. They show that V_2' < V_2'', and that the internal rotation axis
is nearly parallel to the a-axis of the cluster. If it is assumed that the structure
of the rotor part does not change upon electronic excitation, the internal motion
becomes simply a rotation of the water molecule around its symmetry axis. Assuming
this motion, barriers of 180 cm-1 and 130 cm-1 could be estimated for the S0 and
S1 states, respectively. The analysis of the rotational constants of the cluster
yielded an O--O distance of the hydrogen bond of 2.93 angstrom in the ground state
and 2.89 angstrom in the electronically excited state. In the equilibrium structure
of the cluster, the plane containing phenol bisects the plane of the water molecule.(ref)
The rotationally resolved UV excitation spectra of the S1(1Lb)<--S0 origin bands
of 3-methylindole and 5-methylindole have been measured and analyzed. As a result
of an internal rotation of the methyl group, each spectrum consists of rotational
lines of overlapping 0a1<--0a1 and 0e<--0e torsional transitions. Like indole,
3-methylindole and 5-methylindole undergo axis reorientation upon electronic excitation.
The Hamiltonian used to describe all observed spectral includes a pure rotational
part, a pure torsional part, and terms describing the interaction between the
internal rotation and the overall rotation. It also accounts for the axis reorientation
effect. Values for the barrier heights of the methyl torsion, the angle of the
methyl top axis with the inertial axes, and the rotational constants are obtained
for both the S0 and the S1 state. From an analysis of the intensities of the rotational
transitions, the direction of the transition moment and the axis reorientation
angle are obtained. Due to quantum interference effects in the 5-methylindole
spectrum the sign of these angles could be determined. (ref)
The electronic transitions of o-fluorophenol situated at 36799.382 cm-1 and 36906.710
cm-1, denoted the A and B bands, respectively, have been investigated by high
resolution fluorescence excitation spectroscopy. Hole burning studies together
with the high resolution spectroscopy results show that both bands originate in
the same ground state and can be fitted to the rotational constants of the cis
isomer. The rotational constants for the excited states are found to be A = 3231.795
MHz, B = 2207.92 MHz and C = 1313.97 MHz for the A band and A = 3226.945 MHz,
B = 2211.24 MHz and C = 1321.03 MHz for the B band. The planarity of the ground
state is lost upon electronic excitation, which enhances the activity of an out-of-plane
vibration. The A and B band transitions arise from excitations to respectively
the zero and first overtone levels in the double-minimum potential of this out-of-plane
vibration, which shows similarities to the so-called butterfly mode observed in
other benzene derivatives.(ref)
The rotationally resolved fluorescence excitation spectrum of the 0-0 band in
the S1 <-- S0 transition of 4-aminobenzonitrile (ABN) was recorded, at 299
nm, by using laser induced fluorescence in a molecular beam apparatus. This spectrum
exhibits pure b-type character, which indicates that the electronic transition
moment vector is oriented along the short molecular axis. The rotational constants
of the S0 and S1 states were determined. In addition, the rotationally resolved
fluorescence excitation spectra of two vibronic bands in the S1 state, at 807
and 816 cm-1, were recorded. The molecular structure of the ABN molecule is discussed
by comparing the rotational constants and the inertial defects.(ref)
High resolution ultraviolet spectroscopy has been used to investigate the rotationally
resolved excitation spectrum of the first singlet-singlet transition in the benzoic
acid dimer. The measured spectrum consists of two overlapping components. The
corresponding lines in the two components are shown to originate in different
levels of the ground state potential separated by a tunneling splitting produced
by concerted proton exchange between the two subunits forming the dimer. The frequency
separation between the two components is equal to the difference between the tunneling
splittings in the ground and the excited electronic state. This frequency separation
is found to be 1107±7 MHz. From the analysis, it is estimated that the barrier
for proton tunneling changes by about 20% upon electronic excitation. The structure
of the dimer in the ground state is determined to be linear, while in the excited
S1 state it is slightly bent (3.4°±1.7°).(ref)