Structure of phenol, phenol-water and phenol-dimer as determined with spectroscopy
With rotationally resolved electronic spectroscopy, we have determined the structure of phenol, phenol-water, and the phenol-dimer in the electronic ground state and in the first excited singlet state. The measured spectra are perturbed as a result of internal motions: we see the OH of phenol rotating around the C-O bond (so called tunneling of the proton) and we see the water molecule rotating in the phenol-water complex.
The S1 <-- S0 0-0 transitions of phenol and the hydrogen bonded
phenol-water cluster have been studied by high resolution fluorescence
excitation spectroscopy. All lines in the monomer spectrum are split by
56±4 MHz due to the internal rotation of the --OH group about the
a-axis. The barrier for this internal motion is determined in the
ground and excited states; V_2''=1215 cm-1, and V_2'=4710 cm-1. The
rotational constants for the monomer in the ground state are in
agreement with those reported in microwave studies. The excited state
rotational constants were found to be A'=5313.7 MHz, B'=2620.5 MHz, and
C'=1756.08 MHz. The region of the redshifted 0-0 transition of
phenol-water shows two distinct bands which are 0.85 cm-1 apart. Their
splitting arises from a torsional motion which interchanges the two
equivalent H-atoms in the H2O moiety of the cluster. This assignment
was confirmed by spin statistical considerations. Both bands could be
fit to rigid rotor Hamiltonians. Due to the interaction between the
overall rotation of the entire cluster and the internal rotation, both
bands have different rotational constants. They show that V_2' <
V_2'', and that the internal rotation axis is nearly parallel to the
a-axis of the cluster. If it is assumed that the structure of the rotor
part does not change upon electronic excitation, the internal motion
becomes simply a rotation of the water molecule around its symmetry
axis. Assuming this motion, barriers of 180 cm-1 and 130 cm-1 could be
estimated for the S0 and S1 states, respectively. The analysis of the
rotational constants of the cluster yielded an O--O distance of the
hydrogen bond of 2.93 angstrom in the ground state and 2.89 angstrom in
the electronically excited state. In the equilibrium structure of the
cluster, the plane containing phenol bisects the plane of the water
molecule.(ref)
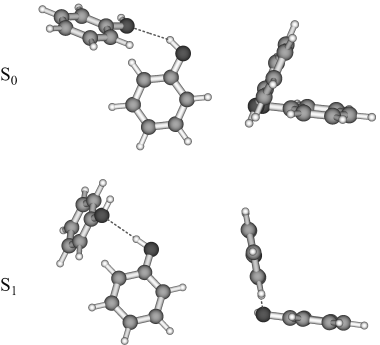
The rotationally resolved UV spectra of the electronic origins of five isotopomers of the phenol dimer have been measured. The complex spectra are analyzed using a fitting strategy based on a genetic algorithm. The intermolecular geometry parameters have been determined from the inertial parameters for both electronic states and compared to the results of ab initio calculations. In the electronic ground state, a larger hydrogen-bond length than in the ab initio calculations is found together with a smaller tilt angle of the aromatic rings, which shows a more pronounced dispersion interaction. In the electronically excited state, the hydrogen-bond length decreases, as has been found for other hydrogen-bonded clusters of phenol, and the two aromatic rings are tilted less toward each other.(ref)
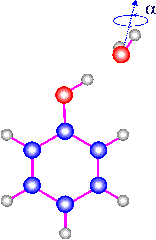 The S1 <-- S0 0-0 transitions of phenol and the hydrogen bonded
phenol-water cluster have been studied by high resolution fluorescence
excitation spectroscopy. All lines in the monomer spectrum are split by
56±4 MHz due to the internal rotation of the --OH group about the
a-axis. The barrier for this internal motion is determined in the
ground and excited states; V_2''=1215 cm-1, and V_2'=4710 cm-1. The
rotational constants for the monomer in the ground state are in
agreement with those reported in microwave studies. The excited state
rotational constants were found to be A'=5313.7 MHz, B'=2620.5 MHz, and
C'=1756.08 MHz. The region of the redshifted 0-0 transition of
phenol-water shows two distinct bands which are 0.85 cm-1 apart. Their
splitting arises from a torsional motion which interchanges the two
equivalent H-atoms in the H2O moiety of the cluster. This assignment
was confirmed by spin statistical considerations. Both bands could be
fit to rigid rotor Hamiltonians. Due to the interaction between the
overall rotation of the entire cluster and the internal rotation, both
bands have different rotational constants. They show that V_2' <
V_2'', and that the internal rotation axis is nearly parallel to the
a-axis of the cluster. If it is assumed that the structure of the rotor
part does not change upon electronic excitation, the internal motion
becomes simply a rotation of the water molecule around its symmetry
axis. Assuming this motion, barriers of 180 cm-1 and 130 cm-1 could be
estimated for the S0 and S1 states, respectively. The analysis of the
rotational constants of the cluster yielded an O--O distance of the
hydrogen bond of 2.93 angstrom in the ground state and 2.89 angstrom in
the electronically excited state. In the equilibrium structure of the
cluster, the plane containing phenol bisects the plane of the water
molecule.(ref)
The S1 <-- S0 0-0 transitions of phenol and the hydrogen bonded
phenol-water cluster have been studied by high resolution fluorescence
excitation spectroscopy. All lines in the monomer spectrum are split by
56±4 MHz due to the internal rotation of the --OH group about the
a-axis. The barrier for this internal motion is determined in the
ground and excited states; V_2''=1215 cm-1, and V_2'=4710 cm-1. The
rotational constants for the monomer in the ground state are in
agreement with those reported in microwave studies. The excited state
rotational constants were found to be A'=5313.7 MHz, B'=2620.5 MHz, and
C'=1756.08 MHz. The region of the redshifted 0-0 transition of
phenol-water shows two distinct bands which are 0.85 cm-1 apart. Their
splitting arises from a torsional motion which interchanges the two
equivalent H-atoms in the H2O moiety of the cluster. This assignment
was confirmed by spin statistical considerations. Both bands could be
fit to rigid rotor Hamiltonians. Due to the interaction between the
overall rotation of the entire cluster and the internal rotation, both
bands have different rotational constants. They show that V_2' <
V_2'', and that the internal rotation axis is nearly parallel to the
a-axis of the cluster. If it is assumed that the structure of the rotor
part does not change upon electronic excitation, the internal motion
becomes simply a rotation of the water molecule around its symmetry
axis. Assuming this motion, barriers of 180 cm-1 and 130 cm-1 could be
estimated for the S0 and S1 states, respectively. The analysis of the
rotational constants of the cluster yielded an O--O distance of the
hydrogen bond of 2.93 angstrom in the ground state and 2.89 angstrom in
the electronically excited state. In the equilibrium structure of the
cluster, the plane containing phenol bisects the plane of the water
molecule.(ref)
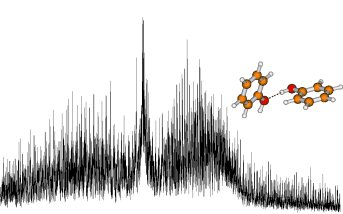
 High resolution UV spectroscopy of phenol and the
hydrogen bonded phenol-water cluster.
High resolution UV spectroscopy of phenol and the
hydrogen bonded phenol-water cluster.